Difference between revisions of "User talk:RobertBuels"
RobertBuels (Talk | contribs) (→Array Representation (ArrayRepr)) |
RobertBuels (Talk | contribs) (→Array Representation (ArrayRepr)) |
||
| Line 145: | Line 145: | ||
Each feature is represented as an array of the form <code>[ class, data, data, ... ]</code>, where the <code>class</code> is an integer index into the track's <code>classes</code> array (more on that in the next section) that refers to the ''array representation'' that defines the meaning of each of the the slots in the array. | Each feature is represented as an array of the form <code>[ class, data, data, ... ]</code>, where the <code>class</code> is an integer index into the track's <code>classes</code> array (more on that in the next section) that refers to the ''array representation'' that defines the meaning of each of the the slots in the array. | ||
| − | An '''array representation''' is encoded in JSON ( | + | An '''array representation''' is encoded in JSON as (didactic comments added): |
{ | { | ||
| − | "attributes": [ | + | "attributes": [ // array of attribute names for this representation |
| − | " | + | "AttributeNameForIndex1", |
| − | " | + | "AttributeNameForIndex2", |
... | ... | ||
], | ], | ||
| − | "isArrayAttr": { | + | "isArrayAttr": { // list of which attributes are themselves arrays |
| − | " | + | "AttributeNameForIndexN": 1, |
... | ... | ||
} | } | ||
Revision as of 16:05, 11 April 2012
Contents
- 1 Rob working on JBrowse docs
- 2 PAGES TO DELETE
- 3 NEW PAGE Template:JBrowseResourcesBoxItem
- 4 NEW PAGE JBrowse
- 5 Demo
- 6 Quick Start: Tutorial
- 7 Installation
- 8 JBrowse Configuration Guide
- 9 Contact
- 10 JBrowse Development
- 11 Presentations
- 12 NEW PAGE JBrowse Advanced Topics
- 13 NEW PAGE JBrowse Troubleshooting
- 14 NEW PAGE JBrowse Configuration Guide
- 15 User Interface
- 16 Reference Sequences
- 17 Feature Tracks
- 18 Image Tracks
- 19 Searchable Names
- 20 Removing Tracks
- 21 URL Control
- 22 See also
- 23 External Links
Rob working on JBrowse docs
This is essentially a branch of the docs. Having to do this is making me wonder whether a wiki is really the best place for them.
PAGES TO DELETE
- JBrowseDev/Installation
- mostly out-of-data CPAN methods rendered unnecessary by setup.sh and bundling of cpanm
NEW PAGE Template:JBrowseResourcesBoxItem
Home Page
Tutorial
Blog
Configuration
Demo
Mailing Lists
NEW PAGE JBrowse
- Mature release
- Active development
- Active support
JBrowse is a genome browser with a fully dynamic AJAX interface. It is very fast and scales well to large datasets. JBrowse renders most tracks entirely within the user's web browser. JBrowse is being developed as the eventual official successor to GBrowse.
Demo
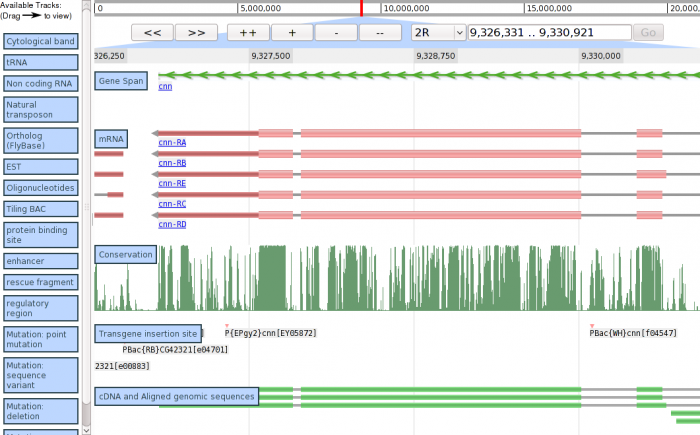
Quick Start: Tutorial
The Getting Started with JBrowse Tutorial provides a basic step-by-step recipe for quickly getting up and running with JBrowse.
Installation
1. Download JBrowse onto your web server.
2. Unpack JBrowse into a directory that is served by your web browser. On many systems, this defaults to /var/www.
cd /var/www tar -xzf jbrowse-*.tar.gz
3. Run the automated-setup script, which will attempt to install all of JBrowse's (modest) prerequisites for you. For the installation to completely succeed, including support for WIG and BAM data, your system must have:
- a working internet connection for downloading Perl modules from CPAN
- compilers for C and C++
-
make - the
svnSubversion client (for fetching thesamtoolscode, needed for BAM support) - development libraries and header files (not just compiled shared objects) for:
- libpng
- Zlib
- GD
For tips on installing these baseline libraries, see #Troubleshooting.
4. When you can see the included Volvox example data, you are ready to configure JBrowse to show your own data! Proceed to the JBrowse Configuration Guide.
JBrowse Configuration Guide
- Configuration Guide
- Formatting reference sequences (e.g. from FASTA files, or a Chado database)
- Creating feature tracks (e.g. from BED or GFF files, a Chado database, or the UCSC genome browser)
- Creating image tracks (e.g. from WIG files)
- Making features searchable by name
- Removing tracks
- Additional topics:
Contact
Please direct questions and inquiries regarding JBrowse to the mailing lists below.
In particular, requests for help should be directed to gmod-ajax@lists.sourceforge.net.
| Mailing List Link | Description | Archive(s) | |
|---|---|---|---|
| JBrowse | gmod-ajax | JBrowse help and general questions. | Nabble (2010/05+), Sourceforge |
| jbrowse-dev | JBrowse development discussions. | Nabble (2011/08+) |
JBrowse Development
JBrowse is an open-source project, started and managed by the laboratory of Ian Holmes at the University of California, Berkeley.
The JBrowse source code repository is kept on GitHub. Please feel very free to fork the code on GitHub and make modifications and improvements, submitting pull requests. GitHub has a very nice tutorial on how to get started with this style of development.
As of January 2012, the lead developer of JBrowse is Robert Buels. Most of JBrowse was originally written by Mitch Skinner.
There is a mailing list for developers, and there is usually a teleconference on the 3rd Monday of the month at 2pm Pacific US time. We welcome participation from Please contact Robert Buels if you would like to listen in or participate.
Presentations
| Date | Presenter | Venue | Links |
|---|---|---|---|
| April 2012 | Robert Buels | GMOD 2012 Community Meeting | |
| January 2012 | Robert Buels | Plant and Animal Genome (PAG) XX | |
| April 2010 | Mitch Skinner | UCSC genome browser group ("genecats") meeting | |
| August 2009 | Ian Holmes | GMOD Community Meeting | Talk summary | PDF |
| January 2009 | Mitch Skinner | GMOD Community Meeting | Talk summary | PDF |
| July 2008 | Ian Holmes | GMOD Community Meeting | Talk summary | PDF |
NEW PAGE JBrowse Advanced Topics
Data Format Specification: Lazy Nested Containment List (NCList) Feature Store
JBrowse uses lazily-loaded nested containment lists (LazyNCLists) as an efficient format for storing feature data in pre-generated static files. A nested containment list is a tree data structure in which the nodes of the tree are intervals themselves features, with the leaves being the features that lie within the bounds of (but are not necessarily subfeatures of) that feature. It has some similarities to an R tree. For more on NClists, see the Alekseyenko paper.
The LazyNCList format can actually be broken down into two distinct subformats: the LazyNCList itself, and the array-based JSON representation of the features themselves.
Array Representation (ArrayRepr)
For speed and memory efficiency, JBrowse feature JSON represents features as arrays instead of objects. This is because, while JavaScript arrays actually behave as objects, many browsers significantly optimize the handling of JavaScript arrays over more general objects.
Each feature is represented as an array of the form [ class, data, data, ... ], where the class is an integer index into the track's classes array (more on that in the next section) that refers to the array representation that defines the meaning of each of the the slots in the array.
An array representation is encoded in JSON as (didactic comments added):
{
"attributes": [ // array of attribute names for this representation
"AttributeNameForIndex1",
"AttributeNameForIndex2",
...
],
"isArrayAttr": { // list of which attributes are themselves arrays
"AttributeNameForIndexN": 1,
...
}
}
Lazy Nested-Containment Lists (LazyNCList)
This data format is currently used in JBrowse 1.3 for tracks of type FeatureTrack, and the code that actually reads this format is in SeqFeatureStore/NCList.js and ArrayRepr.js.
Data Format Specification: Fixed-Resolution Tiled Image Store
JBrowse can display tracks composed of precalculated image tiles, stretching the tile images horizontally when necessary. The JBrowse Volvox example data has a wiggle data track that is converted to image tiles using the included wig2png program, but any sort of image tiles can be displayed if they are laid out in this format.
The files for a tiled image track are structured by default like this:
data/tracks/<track_label>/<refseq_name>/trackData.json data/tracks/<track_label>/<refseq_name>/<zoom_level_urlPrefix>/<index>.png ... (and so on, for many more PNG image files)
Where the PNG files are the image tiles themselves, and trackData.json contains metadata about the track in JSON format, including available zoom levels, the width and height of the image tiles, their base resolution (number of reference sequence base pairs per image tile), and statistics about the data (such as the global minimum and maximum of wiggle data).
The structure of the trackData.json file is:
{
"tileWidth": 2000, // width of all image tiles, in pixels
"stats" : { // any statistics about the data being represented
"global_min": 100,
"global_max": 899
},
"zoomLevels" : [ // array describing what resolution levels are available
{ // in the precalculated image tiles
"urlPrefix" : "1/",
"height" : 100,
"basesPerTile" : 2000
},
... (and so on, for zoom levels in order of decreasing resolution / increasing bases per tile )
]
}
To see a working example of this in action, see the contents of sample_data/json/volvox/tracks/volvox_microarray.wig/ctgA after the Volvox wiggle sample data has been formatted.
The code for working with this tiled image format in JBrowse 1.3 is in TiledImageStore/Fixed.js.
NEW PAGE JBrowse Troubleshooting
Installing prerequisites
Linux - Ubuntu / Debian
These commands, or similar, should install what you need:
sudo apt-get install build-essential libpng-dev zlib1g-dev libgd2-xpm-dev
Installing prerequisites under Linux - Red Hat / Fedora / CentOS
These commands, or similar, should install what you need:
sudo yum groupinstall "Development Tools" sudo yum install libpng-devel gd-devel zlib-devel perl-ExtUtils-MakeMaker
Mac OS X
Use MacPorts, Fink, Homebrew, or another package manager to install a C++ compiler, libpng development headers, GD development headers, and Zlib development headers.
Failures of setup.sh
setup.sh creates a log file of debugging information associated with your installation. Email this to entire file to gmod-ajax@lists.sourceforge.net with a request for support.
As more users try setup.sh and report problems to the mailing list, this wiki will be updated with fixes for common problems they encounter.
NEW PAGE JBrowse Configuration Guide
Setting up JBrowse consists of placing a copy of the jbrowse directory somewhere in the web-servable part of your server's file system, and then running several server-side scripts to create a directory containing JSON-format data files that JBrowse uses to display data. Both the JBrowse code and these data files must be in a location where the web server can present them to clients. Then, a user pointing their web browser at the appropriate URL will see the JBrowse interface, including sequence and feature tracks reflecting the data source.
There is a particular order to follow when adding data to JBrowse: reference sequence data should be added first (using prepare-refseqs.pl`), followed by annotation data. Once all of annotation data has been added, it is possible to make the names of each feature searchable. While there is some flexibility in this order of operations (it is possible to add additional reference sequences after feature tracks have been added, for example), the first step will always be to specify a sequence or set of sequences, and the last step will always be to make the named features searchable (assuming it is desired that all feature names are searchable).
User Interface

2. Hidden Tracks: Drag a track to this area to hide it.
3. Window Slider: Resize the viewing field.
4. Scroll Buttons: Click to scroll by a fixed amount at a given zoom level.
5. Viewing Field: Drag a track to this area to make it visible. Depending on the track, some zooming may be necessary.
6. Zoom Buttons: Click to zoom. Per click, the larger buttons zoom more than the smaller buttons.
7. Chromosome Selector: Choose which chromosome to view.
8. Search Bar: Browse to a certain region by searching for a location or feature name.
Reference Sequences
The reference sequences are the sequences whose annotations the browser will view, and which therefore provide a co-ordinate system for all other tracks. At a close enough zoom level, the sequence data itself is visible as a special track; this track is hidden once the individual sequence characters become too small to distinguish.
The exact interpretation of "reference sequence" will depend on how you are using JBrowse; but for a model organism genome database, each reference sequence would typically represent a chromosome (in a perfect assembly) or at least a contig. Before any feature or image tracks can be input to JBrowse, the reference sequence must be taken into consideration. This is handled by the prepare-refseqs.pl script.
prepare-refseqs.pl
This script is used to input sequence data into JBrowse, and must be run prior to the addition of feature tracks or image tracks. The simplest way to use it is with the --fasta option, which uses a single sequence or set of reference sequences from a FASTA file:
bin/prepare-refseqs.pl --fasta <fasta file> [options]
If the file has multiple sequences (e.g. multiple chromosomes), each sequence will become a reference sequence by default. You may switch between these sequences by selecting the sequence of interest via the pull-down menu to the right of the large "zoom in" button.
You may use any alphabet you wish for your sequences (i.e., you are not restricted to the nucleotides A, T, C, and G; any alphanumeric character, as well as several other characters, may be used). Hence, it is possible to browse RNA and protein in addition to DNA. However, some characters should be avoided, because they will cause the sequence to "split" - part of the sequence will be cut off and and continue on the next line. These characters are the hyphen and question mark. Unfortunately, this prevents the use of hyphens to represent gaps in a reference sequence.
In addition to reading from a fasta file, prepare-refseqs.pl can read sequences from a gff file or a database. In order to read fasta sequences from a database, a config file must be used.
Syntax used to import sequences from gff files:
bin/prepare-refseqs.pl --gff <gff file with sequence information> [options]
Syntax used to import sequences with a config file:
bin/prepare-refseqs.pl --conf <config file that references a database with sequence information> --[refs|refid] <reference sequences> [options]
| Option | Value |
|---|---|
| fasta, gff, or conf | Path to the file that JBrowse will use to import sequences. With the fasta and gff options, the sequence information is imported directly from the specified file. With the conf option, the specified config file includes the details necessary to access a database that contains the sequence information. Exactly one of these three options must be used. |
| out | A path to the output directory (default is 'data' in the current directory) |
| seqdir | The directory where the reference sequences are stored (default: <output directory>/seq) |
| noseq | Causes no reference sequence track to be created. This is useful for reducing disk usage. |
| refs | A comma-delimited list of the names of sequences to be imported as reference sequences. This option (or refid) is required when using the conf option. It is not required when the fasta or gff options are used, but it can be useful with these options, since it can be used to select which sequences JBrowse will import. |
| refids | A comma-delimited list of the database identifiers of sequences to be imported as reference sequences. This option is useful when working with a Chado database that contains data from multiple different species, and those species have at least one chromosome with the same name (e.g. chrX). In this case, the desired chromosome cannot be uniquely identified by name, so it is instead identified by ID. This ID can be found in the 'feature_id' column of 'feature' table in a Chado database. |
Feature Tracks
The feature can be used to visualize localized annotations on a sequence, such as the locations of genes, exons, introns, transposons, SNPs and so forth. There are a number of scripts that can be used with various feature track file formats as inputs to JBrowse:
flatfile-to-json.pl
This script inputs a single track into JBrowse. To put multiple tracks into JBrowse, it must be executed repeatedly.
Terminology: A flat file is a database that exists entirely in a single file. For this script, the flat file must be a GFF3, GFF2, or BED file.
Basic syntax:
bin/flatfile-to-json.pl --[gff|gff2|bed] <flat file> --tracklabel <track name> [options]
Hint: flatfile-to-json.pl simplifies the process of inputting a small number of tracks into JBrowse, since it does not use a config file. If you have many tracks, you will probably want to use a config file, because its structure will make the task of editing tracks easier. In that case, the appropriate script will be biodb-to-json.pl.

| Option | Value |
|---|---|
| gff, gff2, or bed | The name of the file that contains the feature data. The names of these options correspond to the file types, with the exception of gff, which uses a GFF3 file instead of a GFF file. Exactly one of these three options must be used. |
| tracklabel | The internal name that JBrowse will give to this feature track. This option requires a value. |
| key | The external, human-readable label seen on the feature track when it is viewed in JBrowse. The value of key defaults to the value of tracklabel. |
| autocomplete | Dictates what the features of the track will be searchable by after running generate-names.pl. This option can be used with the arguments "label", "alias", "all", or "none". By default, "none" is used.
|
| out | A path to the output directory (default is 'data' in the current directory). |
| cssClass | The css class that will be used to create the feature track. This option makes it possible to choose how the feature track will look by selecting a template class from genome.css. The default css class is 'feature'. |
| getType | Causes the 'type' to be included in the output JSON file. The type is the feature that has been predicted (e.g. promoter site, gene). If a gff file is being used, the type will be in column 3. |
| getPhase | Causes the 'phase' to be included in the output JSON file. The phase describes the reading frame of a DNA (or messenger RNA) sequence. If the phase is relevant, it can have the values 0, 1, or 2; otherwise, the value associated with the phase is '.'. If a gff file is being used, the phase will be in column 8. |
| getSubs | Causes subfeature data to be included in the output JSON file. |
| getLabel | Causes the 'Name' attribute associated with each feature to be included the output JSON file. This will cause a textual name to appear below the features in the track. If a gff3 file is being used, the 'Name' attribute will be in column 9 when it is defined. |
| urlTemplate | A url that your browser will visit when you click on a feature in this track. This is especially useful if you want to link a feature to a page with more information about that feature. |
| arrowheadClass | When this option is used, directional features will be given an arrowhead. The presence and orientation of the arrowhead for each individual feature will depend on data in the input file. Arrowhead classes are defined in genome.css. There is only one that comes with JBrowse (transcript-arrowhead). |
| subfeatureClasses | The css class(es) that will be used for the subfeatures of a feature track. This option makes it possible to choose how the subfeatures will appear. Any of the classes in genome.css can be used for the subfeatures. This option must be used with getSubs in order for subfeatures to appear. |
| clientConfig | Any visual additions or edits for the main features of the track (not for subfeatures). These edits must be specified in JSON syntax. |
| type | The type of feature that will appear in the feature track. This option is useful when the input file contains features of several different types, and you are interested in only having one type of feature (e.g. only having features that are genes) in the feature track. In gff3 files, the type is in the third column. |
| extraData | Use additional information from the input file to create variations in the appearance or behavior of individual features. This option is meant to be used in conjunction with other options. For each feature in the track, a perl subroutine is used to extract additional information, which is then associated with a variable. The value of this variable can be different for each feature. When the name of this variable is surrounded by curly braces and used in the argument for a different option, such as urlTemplate, the feature-specific data is used. |
| nclChunk | The NCList chunk size. This option should not be used unless an error such as "json or perl structure exceeds maximum nesting level" is encountered. If this error does occur, lower the chunk size (the default is 50000). |
bam-to-json.pl
This script inputs a track into JBrowse using a BAM file. Tracks added with this script are similar in appearance to tracks added by flatfile-to-json.pl.
Special dependencies: SAMtools, Bio::DB::SAM
Basic syntax:
bin/bam-to-json.pl --bam <bam file> --tracklabel <track name> [options]
| Option | Value |
|---|---|
| bam | The name of the bam file that contains the feature data. This option requires a value. |
| tracklabel | The internal name that JBrowse will give to this feature track. This option requires a value. |
| key | The external, human-readable label seen on the feature track when it is viewed in JBrowse. The value of key defaults to the value of tracklabel. |
| out | A path to the output directory (default is 'data' in the current directory). |
| cssClass | The css class that will be used to create the feature track. This option makes it possible to choose how the feature track will look by selecting a template class from genome.css. The default css class is 'feature'. |
| clientConfig | Any visual additions or edits for the main features of the track (not for subfeatures). These edits must be specified in JSON syntax. |
| nclChunk | The NCList chunk size in bytes. This option should not be used unless an error such as "json or perl structure exceeds maximum nesting level" is encountered. If this error does occur, lower the chunk size (the default is 50000 bytes). |
| compress | This option causes the output JSON files for the track (trackData.json and hist-*.json) to be compressed with gzip. |
biodb-to-json.pl
This script uses a config file to produce a set of feature tracks in JBrowse. It can be used to obtain information from any database with appropriate schema, or from flat files. Because it can produce several feature tracks in a single execution, it is useful for large-scale feature data entry into JBrowse.
Basic syntax:
bin/biodb-to-json.pl --conf <config file> [options]
| Option | Value |
|---|---|
| conf | The name of the JSON configuration file that will be used. This option must be specified. |
| out | A path to the output directory (default is 'data' in the current directory). |
| track | The identifier of a single track that will be updated or added to JBrowse. In the list of key-value pairs comprising an individual track definition in the config file, the identifier will be the value associated with "track". |
| ref | A comma-delimited list of reference sequence names, used to limit database queries to a subset of JBrowse reference sequences. By default, the database is queried for all reference sequences in JBrowse. |
| refid | A comma-delimited list of reference sequence IDs from a Chado database, used to limit database queries to a subset of JBrowse reference sequences. By default, the database is queried for all reference sequences in JBrowse. |
| compress | This option causes the output JSON files for the track (trackData.json and hist-*.json) to be compressed with gzip. |
ucsc-to-json.pl
This script uses data from UCSC genome annotation database. To reach this data, go to hgdownload.cse.ucsc.edu and click the link for the genome of interest. Next, click the "Annotation Database" link. The data relevant to ucsc-to-json.pl (*.sql and *.txt.gz files) can be downloaded from either this page or the FTP server described on this page.
Together, a *.sql and *.txt.gz pair of files (such as cytoBandIdeo.txt.gz and cytoBandIdeo.sql) constitute a database table. Ucsc-to-json.pl uses the *.sql file to get the column labels, and it uses the *.txt.gz file to get the data for each row of the table. For the example pair of files above, the name of the database table is "cytoBandIdeo". This will become the name of the JBrowse track that is produced from the data in the table.
In addition to all of the feature-containing tables that you want to use as JBrowse tracks, you will also need to download the trackDb.sql and trackDb.txt.gz files for the organism of interest.
Basic syntax:
bin/ucsc-to-json.pl --in <directory with files from UCSC> --track <database table name> [options]
Hint: If you're using this approach, it might be convenient to also download the sequence(s) from UCSC. These are usually available from the "Data set by chromosome" link for the particular genome or from the FTP server.
| Option | Value |
|---|---|
| in | A directory containing all of the *.sql and *.txt.gz data from UCSC. This directory must contain the trackDb.sql and trackDb.txt.gz files for the organism of interest, as well as all of the feature-containing tables that you wish to use as JBrowse tracks. |
| track | The name of the database table. If you leave off the .sql or .txt.gz extensions of the table files you wish to use, you will have this value. |
| out | A path to the output directory (default is 'data' in the current directory). |
| cssClass | The css class that will be used to create the feature track. This option makes it possible to choose how the feature track will look by selecting a template class from genome.css. The default css class is 'feature'. |
| arrowheadClass | When this option is used, directional features will be given an arrowhead. The presence and orientation of the arrowhead for each individual feature will depend on data in the input file. Arrowhead classes are defined in genome.css. There is only one that comes with JBrowse (transcript-arrowhead). |
| subfeatureClasses | The css class(es) that will be used for the subfeatures of a feature track. This option makes it possible to choose how the subfeatures will appear. Any of the classes in genome.css can be used for the subfeatures. |
| clientConfig | Any visual additions or edits for the main features of the track (not for subfeatures). These edits must be specified in JSON syntax. |
| nclChunk | The NCList chunk size in bytes. This option should not be used unless an error such as "json or perl structure exceeds maximum nesting level" is encountered. If this error does occur, lower the chunk size (the default is 50000 bytes). |
| compress | This option causes some of the output JSON files (trackData.json and hist-*.json) to be compressed with gzip. |
| sortMem | The maximum amount of RAM (in bytes) to use for sorting the features. The default value is 536870912 bytes (512MiB). |
Image Tracks
As well as feature tracks, JBrowse allows generic "image tracks". These currently include "quantitative tracks" (which are pixel-resolution histograms) and "basepair tracks" (an experimental track type that shows base-pair arcs in RNA structure, and is intended to demonstrate the Perl API for rendering your own tracks from data).
wig-to-json.pl
Using a WIG file, this script inputs a single wiggle track into JBrowse. In a wiggle track, a numeric value is associated with each nucleotide position in the reference sequence. This is represented in JBrowse as a track that looks like a histogram, where the horizontal axis is for each nucleotide position, and the vertical axis is for the number associated with that position. The vertical axis currently does not have a scale; rather, the heights for each position are relative to each other.
Special dependencies: libpng
In order to use wig-to-json.pl, the code for wig2png must be compiled. This can be done with the following command:
make
Note: If you are using Mac OS X, it might be necessary to execute 'make' in the following way:
make GCC_LIB_ARGS=-L/usr/X11/lib GCC_INC_ARGS=-I/usr/X11/include
Basic syntax:
bin/wig-to-json.pl --wig <wig file> --tracklabel <track name> [options]
Hint: If you are using this type of track to plot a measure of a prediction's quality, where the range of possible quality scores is from some lowerbound to some upperbound (for instance, between 0 and 1), you can specify these bounds with the max and min options.

| Option | Value |
|---|---|
| wig | The name of the wig file that will be used. This option must be specified. |
| tracklabel | The internal name that JBrowse will give to this feature track. This option requires a value. |
| key | The external, human-readable label seen on the feature track when it is viewed in JBrowse. The value of key defaults to the value of tracklabel. |
| out | A path to the output directory (default is 'data' in the current directory). |
| tile | The directory where the tiles, or images corresponding to each zoom level of the track, are stored. Defaults to data/tiles. |
| bgcolor | The color of the track background. Specified as "RED,GREEN,BLUE" in base ten numbers between 0 and 255. Defaults to "255,255,255". |
| fgcolor | The color of the track foreground (i.e. the vertical bars of the wiggle track). Specified as "RED,GREEN,BLUE" in base ten numbers between 0 and 255. Defaults to "105,155,111". |
| width | The width in pixels of each tile. The default value is 2000. |
| height | The height in pixels of each tile. Changing this parameter will cause a corresponding change in the top-to-bottom height of the track in JBrowse. The default value is 100. |
| min | The lowerbound to use for the track. By default, this is the lowest value in the wiggle file. |
| max | The upperbound to use for the track. By default, this will be the highest value in the wiggle file. |
draw-basepair-track.pl
This script inputs a single base pairing track into JBrowse. A base pairing track is a distinctive track type that represents base pairing between nucleotides as arcs.
Basic syntax:
bin/draw-basepair-track.pl --gff <gff file> --tracklabel <track name> [options]

| Option | Value |
|---|---|
| gff | The name of the gff file that will be used. This option must be specified. |
| tracklabel | The internal name that JBrowse will give to this feature track. This option requires a value. |
| key | The external, human-readable label seen on the feature track when it is viewed in JBrowse. The value of key defaults to the value of tracklabel. |
| out | A path to the output directory (default is 'data' in the current directory). |
| tile | The directory where the tiles, or images corresponding to each zoom level of the track, are stored. Defaults to data/tiles. |
| bgcolor | The color of the track background. Specified as "RED,GREEN,BLUE" in base ten numbers between 0 and 255. Defaults to "255,255,255". |
| fgcolor | The color of the track foreground (i.e. the base pairing arcs). Specified as "RED,GREEN,BLUE" in base ten numbers between 0 and 255. Defaults to "0,255,0". |
| width | The width in pixels of each tile. The default value is 2000. |
| height | The height in pixels of each tile. Changing this parameter will cause a corresponding change in the top-to-bottom height of the track in JBrowse. The default value is 100. |
| thickness | The thickness of the base pairing arcs in the track. The default value is 2. |
| nolinks | Disables use of file system links to compress duplicate image files. |
Searchable Names
generate-names.pl
This script makes it possible to search for features by label (the visible name below a feature in JBrowse) and/or by alias (a secondary name that is not visible in the web browser, but may be present in the JSON used by the JBrowse client). For tracks that are added using flatfile-to-json.pl or biodb-to-json.pl, searchability depends on how the 'autocomplete' option is used. If a track is input with the autocomplete option set to 'alias', for instance, features will be searchable by alias after generate-names.pl is run (provided that alias names are present in the original data source). For tracks added using ucsc-to-json.pl, features will be searchable by label after running generate-names.pl.
To search for a term, use the text box at the top of the JBrowse window.
Basic syntax:
bin/generate-names.pl [options]
Note that generate-names.pl does not require any arguments. However, some options are available:
| Option | Value |
|---|---|
| dir | A path to the output directory (default is 'data/names' in the current directory). |
| thresh | A lower-bound on the Patricia trie chunk size. Specifically, the lowest possible chunk size is (thresh + 1). The default value is 200. In this context, a chunk is a group of connected Patricia trie nodes that can be visualized as a single entity, and the chunk size is the total number of genomic features contained in a chunk. The lower the value of thresh, the more chunks there will be. |
| verbose | This setting causes information about the division of nodes into chunks to be printed to the screen. |
Removing Tracks
Although JBrowse does not support a script that removes individual tracks, there are a number of possible options that can be used to change or remove a track:
1. Overwrite the unwanted track with a new track. This is useful when a mistake was made in preparing a track, and you are interested in removing the track only so that you can replace it with a correct track that has the same tracklabel (the 'tracklabel' is a track's internal name). To overwrite a track, use new information with the same tracklabel value.
2. Remove the entire data directory. This is useful when you want to completely remove a track (i.e., remove a tracklabel). This is the easiest and fastest way to remove a track, if replacing all of the correct tracks is trivially simple.
3. Remove the information about the specific tracks from the data directory. This is the most precise option, but it is not always the easiest or best option. In order to delete a specific track while leaving all other tracks intact, it will be necessary to find the entry for the track in the 'data/trackInfo.js' file and remove it. The entry will be enclosed in curly braces, and will contain the track's label and key, in addition to other data. Remove the entire entry, including the curly braces. To adhere to JSON syntax, be sure to also remove any trailing commas. This will completely remove the track from view in JBrowse. However, the track's data will still be present. In order to remove it, remove the 'data/tracks/<reference sequence name>/<tracklabel>/' directory. You might also want to remove the 'data/names' directory and run generate-names.pl again, if any features from the track you removed were searchable.
URL Control
JBrowse provides a number of option for changing the current view in the browser by adding options to the URL which potentially contain genomic location components.
Basic syntax:
http://<server>/<path to jbrowse>?loc=<location string>&tracks=<tracks to show>
loc
Parameters represent the current genomic position which will be visible in the viewing field. Possible input structures are:
"Chromosome"+":"+ start point + ".." + end point
A chromosome name/ID followed by “:”, starting position, “..” and end position of the genome to be viewed in the browser is used as an input. Chromosome ID can be either a string or a mix of string and numbers. “CHR” to indicate chromosome may or may not be used. Strings are not case-sensitive. If the chromosome ID is found in the database reference sequence (RefSeq), the chromosome will be shown from the starting position to the end position given in URL.
example) ctgA:100..200
Chromosome ctgA will be displayed from position 100 to 200.
OR start point + ".." + end point
A string of numerical value, “..” and another numerical value is given with the loc option. JBrowse navigates through the currently selected chromosome from the first numerical value, start point, to the second numerical value, end point.
example) 200..600
OR center base
If only one numerical value is given as an input, JBrowse treats the input as the center position. Then an arbitrary region of the currently selected gene is displayed in the viewing field with the given input position as the center base.
example) 200
OR feature name/ID
If a string or a mix of string and numbers are entered as an input, JBrowser treats the input as a feature name/ID of a gene. If the ID exists in the database RefSeq, JBrowser displays an arbitrary region of the feature from the the position 0, starting position of the gene, to a certain end point.
example) ctgA
tracks
parameters are comma-delimited strings containing track names, each of which should correspond to the "label" element of the track information dictionaries that are currently viewed in the viewing field. Names for the tracks can be found in data/trackInfo.js in jbrowse-1.2.1 folder.
example) DNA,knownGene,ccdsGene,snp131,pgWatson,simpleRepeat
See also
- Installation notes for Mac OS X and Linux
- Using a database with JBrowse
- Using configuration files
- JBrowse Tutorial