NOTE: We are working on migrating this site away from MediaWiki, so editing pages will be disabled for now.
InterMine Presentation
From GMOD
This Wiki page is an edited version of Gos's presentation
Contents
Background
InterMine was developed as the generic underpinnings of the FlyMine Project
- Team of 7 FTE
- 5 developers, one sys admin,
- 1 biologist/ bioinformatician
- Java/ postgreSQL
- SVN repository: 125,000 lines of code + 57,000 lines of tests
- Under development since 2002
- In use by others in Cambridge, Edinburgh, Vienna… + modENCODE DCC if funded
- modENCODE/ Chado
Technical Overview
- Data model --> Java classes, relational schema, mappings through automatic code generation
- Custom Java object/relational system
- When we started, couldn’t select from multiple classes at one time using hibernate.
- Optimised for read-only performance
- Designed for big, complex queries, bulk data
- Performance optimisation
- Transparent query re-writing
- Web application - Struts/JSP/Ajax
Loading Data
- Read-only in production environment (therefore Problems 3 and 5 skipped)
- Load data from InterMine XML
- Parsers from standard formats
- e.g. UniProt, GFF3, PSI, FASTA
- Powerful integration system: coarse/fine grained data source priorities give load-order independence
Test problems
- Used SOFA as core data model - similar to Chado.
- Added Gene.description (absent from model), compiled, loaded data (here XML + FASTA), released webapp.
Example InterMine XML for Problem 1: load genes + annotation
<xml> <items>
<item id="0_3" class=”” implements="http://www.flymine.org/model/genomic#Gene"> <attribute name="identifier" value="xfile" /> <attribute name="description" value="A test gene for GMOD meeting" /> <reference name="organism" ref_id="0_1" /> <collection name="transcripts"> <reference ref_id="0_9" /> </collection> </item> <item id="0_1" class="" implements="http://www.flymine.org/model/genomic#Organism"> <attribute name="taxonId" value="7227" /> </item> ...
</xml>
Resulting webapp object page
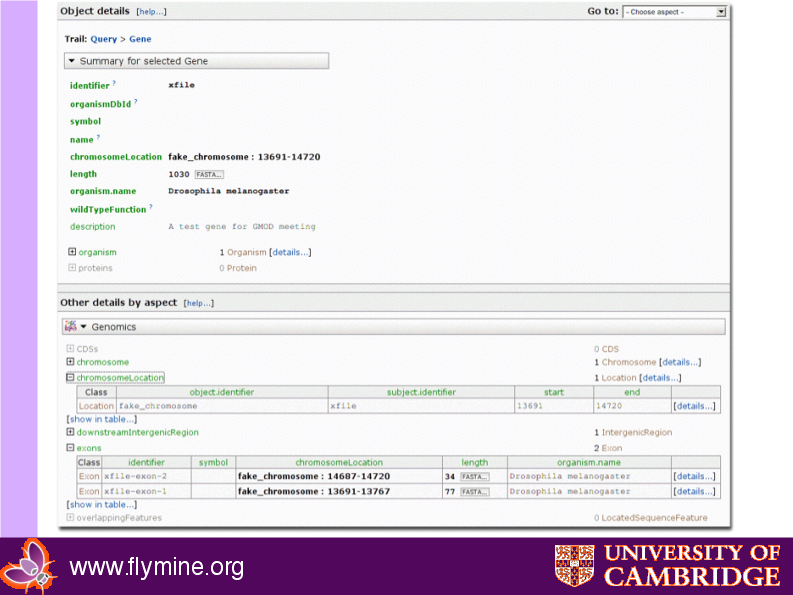{kind=link}
Code for Problem 2: Print gene annotation report
<java> public class BakeOff {
public static void main(String[] args) throws Exception {
// code to get the "xfile" gene
ObjectStore os = ObjectStoreFactory.getObjectStore("os.production");
Query q = new Query();
QueryClass qcObj = new QueryClass(Gene.class);
q.addFrom(qcObj);
QueryField qf = new QueryField(qcObj, "identifier");
q.addToSelect(qf);
SimpleConstraint sc = new SimpleConstraint(qf, ConstraintOp.EQUALS, new QueryValue("xfile"));
q.setConstraint(sc);
System.err.println("query: " + q);
Results res = os.execute(q);
// a Results object is a List of Lists
List rr = (List) res.get(0);
Gene gene = (Gene) rr.get(0);
System.err.println ("symbol: " + gene.getIdentifier());
// a BioEntity in FlyMine has a collection of Synonym objects -
// we need Synonym.value for each Synonym
System.err.print ("synonyms: ");
Iterator synIter = gene.getSynonyms().iterator();
while (synIter.hasNext()) {
Synonym syn = (Synonym) synIter.next();
System.err.print (syn.getValue() + ' ');
}
System.err.println ("description: " + gene.getDescription());
// get the class name, but we already know that the gene is a Gene
System.err.println ("type: " + gene.getClass().getName());
// make a List from a the Set of exons for this Gene
List exons = new ArrayList(gene.getExons());
Exon exon1 = (Exon) exons.get(0);
Exon exon2 = (Exon) exons.get(1);
// get the start and end via the Location object
System.err.println ("exon1 start: " + exon1.getChromosomeLocation().getStart());
System.err.println ("exon1 end: " + exon1.getChromosomeLocation().getEnd());
System.err.println ("exon2 start: " + exon2.getChromosomeLocation().getStart());
System.err.println ("exon2 end: " + exon2.getChromosomeLocation().getEnd());
// write out the first cds
List cdss = new ArrayList(gene.getCDSs());
FlyMineSequence flymineSequence = FlyMineSequenceFactory.make((CDS) cdss.get(0));
// use BioJava to output the sequence
Annotation annotation = flymineSequence.getAnnotation();
annotation.setProperty(FastaFormat.PROPERTY_DESCRIPTIONLINE,
gene.getIdentifier() + " cds");
SeqIOTools.writeFasta(System.err, flymineSequence);
}
}
</java>
Quicksearch - Problem 4: find genes starting with x
Java API
<java>
Query q = new Query(); QueryClass qcObj = new QueryClass(Gene.class); q.addFrom(qcObj); q.addToSelect(qcObj);
QueryField qf = new QueryField(qcObj, "identifier");
SimpleConstraint sc = new SimpleConstraint(qf, ConstraintOp.MATCHES, new QueryValue("x-%"));
q.setConstraint(sc);
</java>
IQL
<sql>
SELECT DISTINCT a1_.identifier AS a2_ FROM org.flymine.model.genomic.Gene AS a1_ WHERE a1_.identifier LIKE 'x-%'
</sql>
Perl API
<perl>
my $genes = InterMine::Gene::Manager->get_genes(query => [
identifier => { like => 'x-%' },],);
</perl>
Larger Query
Within FlyMine: For one or more genes report:
- Gene, Transcripts, Exons, Chromosomal Locations, Lengths
- Query joins 7 classes
- all are on select list of query
- many more tables than classes are joined
- Performance:
- One gene:
- 2 rows in ~2 seconds
- All genes, all organisms
- ~300,000 rows in 36 seconds (without using pre-computation to enhance performance)
- ~300,000 rows in ~1 second (using pre-computation)
- One gene:
Implications of Query Optimisation
- Performance optimisation not tied to schema design
- Can adapt performance optimisation to usage of live database
- Template queries pre-computed
- ~40 template queries run per gene details page - renders in seconds
Acknowlegements
- Richard Smith
- Kim Rutherford
- Matthew Wakeling
- Xavier Watkins
- Julie Sullivan
- Rachel Lyne
- Hilde Janssens
- François Guillier
- Philip North
- Tom Riley
- Peter Mclaren
- Mark Woodbridge
- Debashis Rana
- Wenyan Ji
- Markus Brosch
- Florian Reising
- Andrew Varley
- Gos Micklem
InterMine/FlyMine are funded by the Wellcome Trust (grant no. 067205), awarded to M. Ashburner, G. Micklem, S. Russell, K. Lilley and K. Mizuguchi.