GMOD
JBrowse Tutorial 2010

This JBrowse tutorial was presented by Mitch Skinner at the 2010 GMOD Summer School - Americas, May 2010. The most recent JBrowse tutorial can be found at the JBrowse Tutorial page.
This tutorial walks you through setting up and running a JBrowse server.
Contents
- 1 VMware
- 2 Caveats
- 3 Prerequisites
- 4 JBrowse Introduction
- 5 JBrowse arch
- 6 Setting up JBrowse
- 7 See also
- 8 Other links
VMware
This tutorial was taught using a VMware system image as a starting point. If you want to start with the same system, download and install the start image (below). See VMware for what software you need to use a VMware system image and for directions on how to get the image up and running on your machine.
Download the [start image and the end image.
Logins:
| Purpose | Username | Password |
|---|---|---|
| Shell | gmod | gmodamericas2010 |
| MySQL | root | gmodamericas2010 |
Caveats
Important Note
This tutorial describes the world as it existed on the day the tutorial was given. Please be aware that things like CPAN modules, Java libraries, and Linux packages change over time, and that the instructions in the tutorial will slowly drift over time. Newer versions of tutorials will be posted as they become available.
Prerequisites
These have already been set up on the VM image.
Perl:
- BioPerl 1.6
- JSON
- JSON::XS (optional, for speed)
System packages:
- libpng12-0
- libpng12-dev
Optional, for BAM files:
- samtools, and its dependency libncurses5-dev
- perl module: Bio::DB::SAM
And this is how they were installed: (don’t do this, this has already been done in the VM)
$ sudo apt-get install git-core libpng12-0 libpng12-dev libncurses5-dev
$ cd ~/Documents/Software
$ wget http://sourceforge.net/projects/samtools/files/samtools/0.1.7/samtools-0.1.7a.tar.bz2/download
$ tar xjf samtools-0.1.7a.tar.bz2
$ cd samtools-0.1.7a/
$ make
$ sudo cpan
cpan[1]> install Bio::DB::Das::Chado Bio::DB::Sam JSON JSON::XS
Also: make sure you can Copy/paste from wiki.
Shell tricks:
- Tab completion
- History
- History search
JBrowse Introduction
How and why JBrowse is different from most other web-based genome browsers, including GBrowse.
More detail: paper
Media:GMODCourse2010-JBrowseIntro.pdf
JBrowse arch
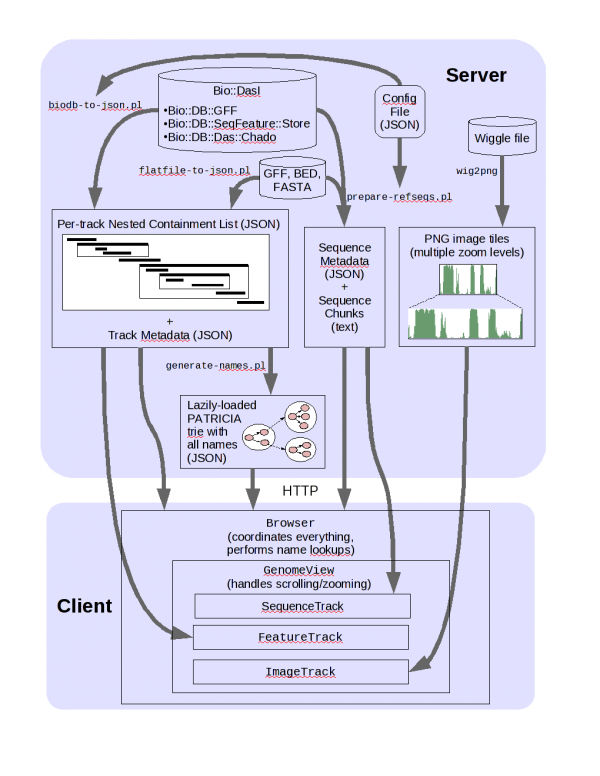
Setting up JBrowse
Getting JBrowse
This has already been done in the VMware image.
- Git
$ sudo apt-get install git-core
$ cd /var/www
$ sudo mkdir jbrowse
$ sudo chown gmod.gmod jbrowse
$ git clone http://github.com/jbrowse/jbrowse.git jbrowse
$ cd jbrowse
$ git branch --track lazyfeatures origin/lazyfeatures
$ git checkout lazyfeatures
Starting Point
Visit in web browser:
- if your web browser is in the VM: http://localhost/jbrowse/
- if your web browser is outside the VM, use your VM’s IP address instead of “localhost”
You should see just a blank white page.
Basic Steps
Setting up a JBrowse instance with feature data goes in three basic steps:
- Specify reference sequences
- Load feature data
- Collect feature names
Data from a database
Here, we’ll use the Chado adapter; other common database adapters are Bio::DB::SeqFeature::Store and Bio::DB::GFF.
Starting config file: ~/Documents/Data/jbrowse/first-config.json
{
"description": "Pythium",
"db_adaptor": "Bio::DB::Das::Chado",
"db_args": { "-dsn": "dbi:Pg:dbname=chado",
"-user": "gmod",
"-pass": ""},
...
Specify reference sequences
The first script to run is bin/prepare-refseqs.pl; that script is the
way you tell JBrowse about what your reference sequences are. Running
bin/prepare-refseqs.pl also sets up the “DNA” track.
Run this from within the /var/www/jbrowse directory (you could run it
elsewhere, but you’d have to explicitly specify the location of the data
directory on the command line).
$ cd /var/www/jbrowse
$ bin/prepare-refseqs.pl --conf ~/Documents/Data/jbrowse/first-config.json --refs scf1117875582023
Visit in web browser: you should new see the JBrowse UI (and if you zoom all the way in, some sequence)
Load Feature Data
Next, we’ll use biodb-to-json.pl to get feature data out of the
database and turn it into JSON data that the
web browser can use.
Add a basic track definition; this will tell biodb-to-json.pl what
features to put into the track, and how the track should look:
...
"TRACK DEFAULTS": {
"class": "feature"
},
"tracks": [
{
"track": "gene",
"key": "Gene",
"feature": ["gene"],
"autocomplete": "all",
"class": "feature2",
"urlTemplate": "http://www.google.com/search?q={name}"
}
]
}
"class" specifies the CSS class that
describes how the feature should look. The classes are specified in the
genome.css file:
$ less genome.css
For this particular track, I’ve specified the "feature2" class which
looks like this in the CSS file:
.plus-feature2,
.minus-feature2 {
position:absolute;
height: 15px;
background-repeat: repeat-x;
cursor: pointer;
min-width: 1px;
z-index: 10;
}
.plus-feature2 { background-image: url('img/plus-herringbone16.png'); }
.minus-feature2 { background-image: url('img/minus-herringbone16.png'); }
Run the bin/biodb-to-json.pl script with this config file to set up
this track:
$ bin/biodb-to-json.pl --conf ~/Documents/Data/jbrowse/first-config.json
(visit in web browser: you should see a new gene track)
More complex track
Now we’ll add a second track; this one will have subfeatures. This
snippet is from: ~/Documents/Data/jbrowse/second-config.json
...
{
"track": "match",
"key": "Matches",
"feature": ["match"],
"autocomplete": "all",
"subfeatures": true,
"class": "generic_parent",
"subfeature_classes": {
"match_part": "match_part"
},
"clientConfig": {
"subfeatureScale": 20
}
}
...
$ bin/biodb-to-json.pl --conf ~/Documents/Data/jbrowse/second-config.json
(visit in web browser: you should see a new track, which has subfeatures if you’re zoomed in far enough)
Collect feature names
When you generate JSON for a track, if you specify "autocomplete" then
a listing of all of the names/IDs from that track (along with the
locations of the corresponding features) will also be generated.
The bin/generate-names.pl script collects those lists of names from
all the tracks and combines them into one big tree that the client uses
to search.
$ bin/generate-names.pl -v
Visit in web browser, search for feature name: e.g.,
maker-scf1117875582023-snap-gene-0.3
Data from flat files
First, remove the data directory:
$ rm -r data
Visit in web browser, see blank screen again
Sequences
You can also get data into JBrowse from flat files. For sequence, use
prepare-refseqs.pl with the --fasta argument:
$ bin/prepare-refseqs.pl --fasta ~/Documents/Data/jbrowse/scf1117875582023.fasta
Visit in web browser; you should see a second reference sequence.
Features
To get feature data from flat files into JBrowse, use
flatfile-to-json.pl. We’ll use some more of the data from the
MAKER session:
$ bin/flatfile-to-json.pl \
--gff /home/gmod/Documents/Data/maker/example2_pyu/finished.maker.output/finished_datastore/scf1117875582023/scf1117875582023.gff \
--type match --getSubs --tracklabel "gff_match" --key "GFF match" \
--cssclass generic_parent --subfeatureClasses '{"match_part": "generic_part_a"}'
Visit in web browser; you should see a new “GFF match” track.
BAM data
The “lazyfeatures” branch of JBrowse can generate JSON from a BAM source:
$ bin/flatfile-to-json.pl \
--bam ~/Documents/Data/jbrowse/simulated-sorted.bam \
--cssclass exon --tracklabel BAM_data --key "BAM Data"
Quantitative data
JBrowse can also display quantitative data in the wiggle format. JBrowse processes wiggle files with a C++ program, which you have to compile:
$ make
Now you can process the wiggle file:
$ bin/wig-to-json.pl --wig ~/Documents/Data/jbrowse/pyu.wig \
--tracklabel "coverage_wig" --key "Wiggle Coverage" --min 0 --max 50
Visit in web browser
See also
- Documentation: JBrowseDev/Main
Other links
- Config file ref: http://jbrowse.org/code/jbrowse-master/docs/config.html
- Misc images: http://www.getdropbox.com/gallery/580036/1/gmod-eu?h=e103e0
- DIV test: http://jbrowse.org/test/boatdiv/boat.html
Facts about “JBrowse Tutorial 2010”
| Has topic | JBrowse + |