Difference between revisions of "GBrowse syn PAG tutorial"
(→The database loading script) |
(→The database loading script) |
||
| Line 129: | Line 129: | ||
===The database loading script=== | ===The database loading script=== | ||
Then, we will load the database. | Then, we will load the database. | ||
| − | * We will use the loading script <span class="pops">[http://gmod. | + | * We will use the loading script <span class="pops">[http://gmod.svn.sourceforge.net/viewvc/gmod/Generic-Genome-Browser/branches/stable/bin/gbrowse_syn/load_alignments_msa.pl load_alignments_msa.pl], which will accept clustalw as an input format. |
'''we are using the options:''' | '''we are using the options:''' | ||
-u root -- username is root | -u root -- username is root | ||
Revision as of 12:43, 13 January 2010
__NOTITLE__
This tutorial walks you through how to install and configure the GBrowse_syn comparative genomics viewer. This tutorial was originally taught by Sheldon McKay at the 2009 GMOD Summer School - Europe & Americas. The notes and VMware image used on this page are from the Europe course.
Contents
- 1 VMware
- 2 Caveats
- 3 The Generic Synteny Browser
- 4 Installing GBrowse_syn
- 5 Loading and Configuration of GBrowse_syn
- 6 Optional Advanced Section
VMware
| This tutorial was taught using a VMware system image as a starting point. If you want to start with that same system, download and install the Starting image.
See VMware for what software you need to use a VMware system image, and for directions on how to get the image setup and running on your machine. |
|
Caveats
Important Note
This tutorial describes the world as it existed on the day the tutorial was given. Please be aware that things like CPAN modules, Java libraries, and Linux packages change over time, and that the instructions in the tutorial will slowly drift over time. Newer versions of tutorials will be posted as they become available.
The Generic Synteny Browser

GBrowse_syn, or the Generic Synteny Browser, is a GBrowse-based synteny browser designed to display multiple genomes, with a central reference species compared to two or more additional species. It can be used to view multiple sequence alignment data, synteny or co-linearity data from other sources against genome annotations provided by GBrowse. GBrowse_syn is included with the standard GBrowse package (version 1.69 and later). Working examples can be seen at TAIR, WormBase, and SGN.
Gbrowse_syn Introduction
- View introductory presentation in GBrowse_syn
- It would be a good idea to download or acquire the sample data during the above presentation.
GBrowse_syn Documentation
There is detailed documentation on the GMOD wiki for how to install, configure and use GBrowse_syn. To get started, browse these pages:
- GBrowse_syn overview
- Installation
- Configuration
- Alignment Data
- The user interface
- Presentations and workshops
Whole Genome Alignments
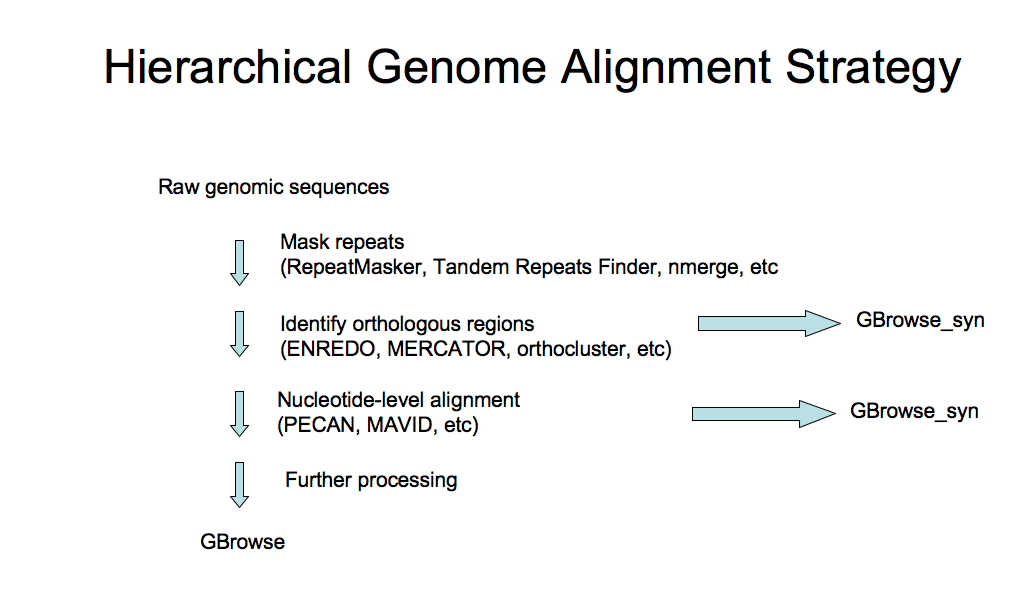
The focus of the section of the course is on dealing with alignment or synteny data and using GBrowse_syn. However, how to generate whole genome alignments, identify orthologous regions, etc, are the subject of considerable interest, so some background reading is listed below:
- Primer on Hierarchical Genome Alignment Strategies
- article on PECAN and ENREDO
- all about PECAN
- The gene annotations for each species are in GFF files.
- The alignment data are in a constrained CLUSTALW format (They were not generated by the program CLUSTALW, which is not necessarily suitable for whole genome alignments)
- There are processing steps for the alignment data but it is very computationally intensive and we will load pre-processed data to get a head start.
Installing GBrowse_syn
GBrowse_syn is part of the GBrowse package and was pre-installed when you went through the GBrowse installation.
Loading and Configuration of GBrowse_syn
The example we will use is a two-species comparison of rice (Oryza sativa) and one of its wild relatives*
*Data courtesy of Bonnie Hurwitz; sequences and names have been obfuscated to protect unpublished data
The instructions for downloading these data to the Ubuntu virtual disk:
$ mkdir ~/data/gbrowse_syn $ cd ~/data/gbrowse_syn $ wget http://mckay.cshl.edu/downloads/rice.tar.bz2 $ tar xjvf rice.tar.bz2
Create a MySQL database
- GBrowse_syn uses a "joining" database to store all of the alignment data
- The first thing we need to do is create a MySQL alignment database using the command-line incantation below:
$ mysql -uroot -e 'create database rice_synteny'
- Then make sure the web user "nobody" can read the database. Pay special attention to the quotes!
$ mysql -uroot -e "GRANT SELECT on rice_synteny.* to 'nobody'@'localhost'"
Loading the alignment data
The alignment data file
Have a look at the input data in clustalw format:
$ cd ~/data/gbrowse_syn/rice
$ more data/rice.aln
CLUSTAL W(1.81) multiple sequence alignment W(1.81)
rice-3(+)/16598648-16600199 ggaggccggccgtctgccatgcgtgagccagacggggcgggccggagacaggccacgtgg
wild_rice-3(+)/14467855-14469373 gggggccgg------------------------------------agacaggccacgtgg
** ****** ***************
rice-3(+)/16598648-16600199 ccctgccccgggctgttgacccactggcacccctgtcccgggttgtcgccctcctttccc
wild_rice-3(+)/14467855-14469373 ccctgccccgggctgttgacccactggcacccctgtcccgggttgtcgccctcctttccc
************************************************************
rice-3(+)/16598648-16600199 cgccatgctctaagtttgctcctcttctcgaacttctctctttgattcttcacgtcctct
wild_rice-3(+)/14467855-14469373 cgccatgctctaagtttgctcctcttctcgaacttctctctttgattcttcacgtcctct
************************************************************
rice-3(+)/16598648-16600199 tggagcctccccttctagctcgatcacgctctgctcttccgcttggaggctggcaaaact
wild_rice-3(+)/14467855-14469373 tggagcctccccttctagctcgatcgcgctctgctcttccgcttggaggctggcaaaact
************************* **********************************
Note on CLUSTALW
These data are in clustalw format. The scripts used to process these data will recognize clustalw and other commonly used formats recognized by BioPerl's AlignIO parser. This does not mean that clustalw is the actual program used to generate the alignment data.
- These particular alignment file in clustalw format was generated using a part of the compara pipeline.
- See this generalized hierarchical whole genome alignment workflow for general information on how whole genome alignment data ca be generated.
{kind=link}
Note on the sequence ID syntax
The sequence ID is this clustal file is overloaded to contain information about the species, strand and coordinates. This information is essential:
rice-3(+)/16598648-16600199 speciesv-refseq(strand)/start-end
The database loading script
Then, we will load the database.
- We will use the loading script load_alignments_msa.pl, which will accept clustalw as an input format.
we are using the options: -u root -- username is root -d rice_synteny -- use database rice_synteny -f clustalw -- use clustalw format -v -- print information about what is happening
other available options that we do not need here:
-p password -- not used because the root user has no password
in this implementation
-n -- do not calculate map coordinates (faster)
-c -- initialize a new (or overwrite the old) database
We will be running the script with this command line incantation (see below):
$ ../bin/load_alignments_msa.pl -u root -d rice_synteny -format clustalw -v data/rice.aln
Running in the background with the linux screen command
Using screen: Running the script as we are below is time-consuming, so we will use a screen session to run it in the background while we turn our attention to downstream tasks. [more information on 'screen'...]
Oops! If you are using the VMware image, you need to first install screen
$ sudo apt-get install screen [sudo] password for gmod: Reading package lists... Done Building dependency tree Reading state information... Done The following NEW packages will be installed: screen 0 upgraded, 1 newly installed, 0 to remove and 33 not upgraded. Need to get 591kB of archives. After this operation, 1004kB of additional disk space will be used. Get:1 http://us.archive.ubuntu.com hardy/main screen 4.0.3-7ubuntu1 [591kB] Fetched 591kB in 1s (452kB/s) Selecting previously deselected package screen. (Reading database ... 33643 files and directories currently installed.) Unpacking screen (from .../screen_4.0.3-7ubuntu1_i386.deb) ... Setting up screen (4.0.3-7ubuntu1) ...
- When entering screen mode, hit 'space' to clear the first screen if a message appears.
- If your backspace key does not work in screen mode, use ^H (ctrl key + H key).
$ cd ~/data/gbrowse_syn/rice $ screen $ ../bin/load_alignments_msa.pl -u root -d rice_synteny -format clustalw -v data/rice.aln Processing alignment file data/rice.aln... Processing alignment 1 Mapping coordinates for alignment 1... Done! Processed pair-wise alignment 1 Processing alignment 2 Mapping coordinates for alignment 2... Done! Processed pair-wise alignment 2 Processing alignment 3 Mapping coordinates for alignment 3... Done! Processed pair-wise alignment 3 Processing alignment 4 Mapping coordinates for alignment 4... Done! Processed pair-wise alignment 4 etc...
- This will go on for some time (there are 1800 alignments), so we will let the screen run in the background and work on our other tasks. We do this like so:
- hit ^A (ctrl key + A key), then release
- hit the D key, which will detach the screen (continues to run in the background)
- We can check back later like so:
$ screen -r
- If the job is done, we can exit the session by typing 'exit' at the command prompt.
What's that error message?
If you see an error message like the one below, there is a harmless bioperl version incompatibility caused by using a version of bioperl that is more recent the the copy on the virtual disk.
--------------------- WARNING --------------------- MSG: Use of method no_sequences() is deprecated, use num_sequences() instead To be removed in 1.0075 ---------------------------------------------------
It does not affect the database loading. You can either:
- ignore the message, or
- edit line 43 of the script to change "$aln->no_sequences" to "$aln->num_sequences"
Setting up the species' databases
GFF3
- Each of the species' databases will be installed in MySQL using the Bio::DB::SeqFeature::Store adapter for GFF3
Let's have a look at the GFF3 data:
$ more rice.gff3 ##gff-version 3 ##sequence-region 3 1 19401704 3 ensembl gene 78 1849 . - . ID=3_FG2548;Name=3_FG2548;biotype=protein_coding 3 ensembl mRNA 78 1849 . - . ID=3_FGT2548;Parent=3_FG2548;Name=3_FGT2548;biotype=protein_coding 3 ensembl CDS 1645 1849 . - 0 Parent=3_FGT2548;Name=CDS.12 3 ensembl CDS 1444 1547 . - 1 Parent=3_FGT2548;Name=CDS.13 3 ensembl CDS 999 1144 . - 0 Parent=3_FGT2548;Name=CDS.14 3 ensembl CDS 799 913 . - 2 Parent=3_FGT2548;Name=CDS.15 3 ensembl CDS 646 786 . - 0 Parent=3_FGT2548;Name=CDS.16 3 ensembl CDS 78 215 . - 0 Parent=3_FGT2548;Name=CDS.17 3 ensembl gene 4910 5518 . + . ID=3_FG2546;Name=3_FG2546;biotype=protein_coding 3 ensembl mRNA 4910 5518 . + . ID=3_FGT2546;Parent=3_FG2546;Name=3_FGT2546;biotype=protein_coding 3 ensembl CDS 4910 5518 . + 0 Parent=3_FGT2546;Name=CDS.19 3 ensembl gene 5743 6351 . - . ID=3_FG2565;Name=3_FG2565;biotype=protein_coding 3 ensembl mRNA 5743 6351 . - . ID=3_FGT2565;Parent=3_FG2565;Name=3_FGT2565;biotype=protein_coding 3 ensembl CDS 5743 6351 . - 0 Parent=3_FGT2565;Name=CDS.21 3 ensembl gene 10979 16914 . + . ID=3_FG2570;Name=3_FG2570;biotype=protein_coding 3 ensembl mRNA 10979 16914 . + . ID=3_FGT2570;Parent=3_FG2570;Name=3_FGT2570;biotype=protein_coding 3 ensembl CDS 10979 11592 . + 0 Parent=3_FGT2570;Name=CDS.29 3 ensembl CDS 11670 13317 . + 2 Parent=3_FGT2570;Name=CDS.30 3 ensembl CDS 13390 14204 . + 0 Parent=3_FGT2570;Name=CDS.31 3 ensembl CDS 14433 16914 . + 2 Parent=3_FGT2570;Name=CDS.32
Some key things to note:
- The ##sequence-region directive
- is used to create a reference sequence named 3, which is the scaffold on which all of the other features in the file are located
- The 'gene' features
- are the top-level parent featured. The 'mRNA' and 'CDS' features are children of the gene. The containement hierarchy is organized using the 'Parent' tag. The CDSs are children of the mRNA, which is in turn a child of the gene. For display purposes, we only need to worry about the gene.
Loading
- Loading the GFF3 into the MySQL database is the same procedure that is used for loading GFF3 databases for GBrowse.
- It uses the bioperl script bp_seqfeature_load.pl.
Note: before we load the GFF3 databases, we need to create a database for each species and give the web user 'nobody' read privileges
$ mysql -uroot Welcome to the MySQL monitor. Commands end with ; or \g. Your MySQL connection id is 13 Server version: 5.0.51a-3ubuntu5.4 (Ubuntu) Type 'help;' or '\h' for help. Type '\c' to clear the buffer. mysql> CREATE DATABASE rice; Query OK, 1 row affected (0.00 sec) mysql> CREATE DATABASE wild_rice; Query OK, 1 row affected (0.00 sec) mysql> GRANT SELECT on rice.* TO 'nobody'@'localhost'; Query OK, 0 rows affected (0.08 sec) mysql> GRANT SELECT on wild_rice.* TO 'nobody'@'localhost'; Query OK, 0 rows affected (0.00 sec) mysql> quit
- Change to the location of the GFF data files
$ cd ~/data/gbrowse_syn/rice/data
- We can use screen to run this script as well to put the loading in the background
- Using the incantation below will cause screen to exit when the job is done, so no need to monitor (though you can if you want to)
- The -f options means "fast load"
- The -c option means complete (or destructive) load. It would overwrite previously loaded 'rice' databases
$ screen bp_seqfeature_load.pl -u root -d rice -c -f rice.gff3 loading rice.gff3... 12000 features loaded in 13.47s ( 1.14s/1000 features)...
- It will run like this for some time.
- Remember, to detach the screen session:
- hit ^A (ctrl key + A key), then release
- hit the D key, which will detach the screen (continues to run in the background)
- In this case we called screen in such a way that it will exit automatically when the job is done.
Repeat for wild rice:
$ screen bp_seqfeature_load.pl -u root -d wild_rice -c -f wild_rice.gff3
Detach this screen and we can move on the the next steps while the databases are loading.
If, at any time you want to see how the database loads are going:
$ screen -r
There are several suitable screens on:
11066.pts-0.gmod (Detached)
11062.pts-0.gmod (Detached)
copy and paste one the IDs (eg: 11066.pts-0.gmod) like so:
$screen -r 11066.pts-0.gmod
and it will show you what's happening with the database load.
If the loading is done, you will get this result:
$ screen -r There is no screen to be resumed.
Setting up the Configuration Files
Copy the configuration file to the installation directory. Note that you will need root privileges to do this.
$ cd /home/gmod/data/gbrowse_syn/rice/conf $ sudo cp rice_synteny.conf wild_rice_synteny.conf oryza.synconf /etc/apache2/gbrowse.conf
A Species Config File
File: rice_synteny.conf
[GENERAL]
description = Domestic rice chromosome 3
db_adaptor = Bio::DB::SeqFeature::Store
db_args = -adaptor DBI::mysql
-dsn dbi:mysql:rice;host=localhost
-user nobody
# examples to show in the introduction
examples = 3:51418..52015
3:67260..67704
# what image widths to offer
image widths = 450 640 800 1024
# default width of detailed view (pixels)
default width = 1024
initial landmark = 3:200000..300000
# Web site configuration info
stylesheet = /gbrowse/gbrowse.css
buttons = /gbrowse/images/buttons
tmpimages = /gbrowse/tmp
# max and default segment sizes for detailed view
max segment = 5000000
default segment = 5000
# zoom levels
zoom levels = 50 100 200 1000 2000 5000 10000 20000 40000 50000 100000 500000 1000000 5000000
# colors of the overview, detailed map and key
overview bgcolor = lightgrey
detailed bgcolor = lightgoldenrodyellow
key bgcolor = beige
default features = EG
balloon tips = 1
[TRACK DEFAULTS]
glyph = generic
height = 10
bgcolor = lightgrey
fgcolor = black
font2color = blue
label density = 25
link = AUTO
link_target = _blank
title = Hello, my name is $name!
################## TRACK CONFIGURATION ####################
# the remainder of the sections configure individual tracks
###########################################################
[EG]
feature = gene:ensembl
glyph = gene
height = 10
bgcolor = peachpuff
fgcolor = hotpink
description = 0
label = 0
category = Transcripts
key = ensembl gene
The GBrowse_syn Config File
File: oryza.synconf
[GENERAL]
description = BLASTZ alignments for Oryza sativa
# The synteny database
join = dbi:mysql:database=rice_synteny;host=localhost;user=nobody
# This option maps the relationship between the species data sources, names and descriptions
# The value for "name" (the first column) is the symbolic name that gbrowse_syn uses to identify each species.
# This value is also used in two other places in the gbrowse_syn configuration:
# 1) the species name in the "examples" directive and the species name in the .aln file
# 2) the species name in the .aln file
# The value for "conf. file" is the basename of the corresponding gbrowse .conf file.
# This value is also used to identify the species configuration stanzas at the bottom of the configuration file.
# name conf. file Description
source_map = rice rice_synteny "Domesic Rice (O. sativa)"
wild_rice wild_rice_synteny "Wild Rice"
tmpimages = /gbrowse/tmp
imagewidth = 800
stylesheet = /gbrowse/gbrowse.css
cache time = 1
config_extension = conf
# example searches to display
examples = rice 3:16050173..16064974
wild_rice 3:1..400000
zoom levels = 5000 10000 25000 50000 100000 200000 400000
# species-specific databases
[rice_synteny]
tracks = EG
color = blue
[wild_rice_synteny]
tracks = EG
color = red
Testing the rice and wild_rice data sources in GBrowse
- If things have worked out, you should see something like the image below when you point you browser to:
http://172.16.109.133/cgi-bin/gbrowse/rice
Note that the IP address will vary and you will use 'localhost' if you are running your browser within the VMware player.

Viewing the data in GBrowse_syn
- Cross you fingers
http://172.16.109.133/cgi-bin/gbrowse_syn/oryza
Note that the IP address will vary

Optional Advanced Section
This optional session will use five pre-built databases.
The instructions for downloading these data to the Ubuntu virtual disk:
$ cd ~/data/gbrowse_syn $ wget ftp://ftp.gmod.org/pub/gmod/Courses/2009/SummerSchoolEurope/nematodes.tar.bz2 $ tar xjvf nematodes.tar.bz2
Deal with the databases (these are MySQL dumps)
$ cd ~/data/gbrowse_syn/nematodes/mysql_dumps
The script load.pl is: <perl>
- !/usr/bin/perl -w
use strict;
while (<*.sql>) {
my ($name) = /(\S+)\.sql/; system "mysql -uroot -e 'drop database $name'"; system "mysql -uroot -e 'create database $name'"; system "mysql -uroot $name <$_"; print "$name loaded\n";
} </perl>
$ ./load.pl &
This will take a while to run. It is building five MySQL databases, two GBrowse_syn data sources (pecan and orthocluster data sets) and one species' database for each of C. elegans, C. remanei and C. briggsae.
NOTE: you will need to give the user 'nobody', as specified in the database connection section of the configuration files, SELECT access to the MySQL databases.
- You must be user 'root' to do this.
- You can use the command-line incantation below:
$ mysql -uroot -e "GRANT SELECT on *.* TO 'nobody'@'localhost'"
Loading Sample Configuration Files
- get the config files
$ cd ~/data/gbrowse/nematodes/conf $ sudo cp * /etc/apache2/gbrowse.conf $ cd /etc/apache2
point your web browser at:
http://localhost:/cgi-bin/gbrowse_syn/orthocluster/
